
Another Attack on the Carve-Out: Novartis Seeks a TRO Enjoining ENTRESTO Generic
August 8, 2024Increasingly the subject of induced infringement litigation, the viability of the carve-out has been questioned for several years now. But recently, a new challenge was filed in the District Court for the District of Columbia questioning whether modifications to labeling as a result of patent protections—beyond the mere omission of language—are permissible under the section viii carve-out requirements. In other words, the case asks how “same” a generic drug’s labeling must be as compared to its Reference Listed Drug.
On July 30, brand name drug sponsor Novartis asked the District Court for a Temporary Restraining Order (here and here) enjoining FDA’s approval of a generic version of its ENTRESTO (sacubitril and valsartan) with certain dosing and indication information carved-out or modified. That modification to the labeling, Novartis argues, “represents a sharp departure from FDA’s statutory and regulatory mandate to require that a generic drug be the ‘same’ as its reference listed drug.” This is because the generic—sponsored by MSN Laboratories Private LTD—revised the approved ENTRESTO indication rather than simply omitted portions. Additionally, Novartis alleged that FDA unlawfully permitted the carve-out of critical safety information of a modified dosing regimen. Each of these issues, says Novartis, “independently renders the agency’s decision unlawful, and invalidates the agency’s approval of the MSN product.”
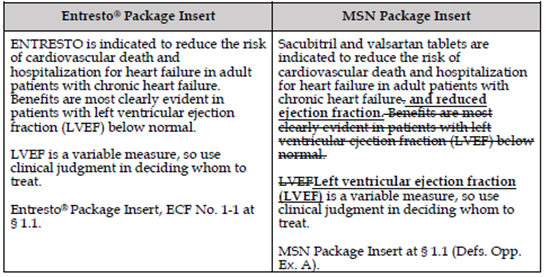
ENTRESTO was approved by FDA in July 2015 “to reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure (NYHA Class II-IV) and reduced ejection fraction.” In February 2021, FDA approved a supplement to the ENTRESTO NDA, which changed the indication such that it now reads “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure. Benefits are most clearly evident in patients with left ventricular ejection fraction (LVEF) below normal.” As a result, ENTRESTO is now approved to treat all patients with chronic heart failure, whether classified as having reduced ejection fraction or not, and added a new dosing regimen for specific patients. Novartis has a patent on the new indication, which claims a modified dosing regimen for use in patients with heart failure with reduced ejection fraction.
While denying a Citizen Petition from Novartis asking FDA to refuse to approve any ANDA that omits the new dosing regimen or changes the indication, FDA approved MSN’s ANDA on July 24, 2024. FDA, in its Citizen Petition Denial, stated that it retains the authority to approve generic labeling that modifies an approved indication and that it could lawfully approve generic labeling that omits the modified dosing regimen in ENTRESTO’s labeling. To that end, FDA approved the MSN ANDA without the modified dosing regimen and with the indication “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure and reduced ejection fraction. Benefits are most clearly evident in patients with left ventricular ejection fraction (LVEF) below normal. Left ventricular ejection fraction (LVEF) is a variable measure, so use clinical judgment in deciding whom to treat.” From the government’s brief, see here.
{kind=link}
![]()
Upon denial of the Citizen Petition and approval of the MSN ANDA, Novartis filed suit against FDA arguing that FDA’s actions violate the plain text of the FDCA. FDA should not have approved the modified indication, as the FDCA requires generic labeling to match the current labeling for the reference listed drug—not the 2015 label that has been discontinued. Novartis further claims that the MSN labeling is unlawful because it violates the statutory requirement that the indications be “the same.” While Novartis implicitly questions whether FDA’s regulations are consistent with the statute that requires the same labeling, it points to FDA regulations that permit only the “omission of an indication”—not the modification of the product’s currently approved indication. Novartis asserts that FDA failed to explain “how a full cloth rewriting of the reference drug’s labeling is consistent with the rest of the ENTRESTO labeling” and that the modified labeling, especially considering the omission of the modified dosing regimen, “renders [the MSN product] both less safe and less effective.”
FDA and intervenor MSN vehemently defend FDA’s approval here. FDA asserts that “there is no legal or logical basis for Novartis’s claim that generic labeling may only omit a patented use by deleting words from (rather than adding them to)” the RLD’s labeling. FDA regulations explicitly permit “omission of an indication or other aspect of labeling protected by patent,” which is exactly what FDA approved here when it carved out the use of patients with normal ejection fraction. FDA further argues that Novartis’s implied preclusion against adding words would lead to absurd results, as the availability of a carve-out would depend on stylistic wording choices in the reference labeling.
Meanwhile, MSN argues that Congress designed the “same labeling” statutory requirements to allow for labeling changes by permitting section viii statements to avoid including language that would infringe a patent. That’s precisely what FDA did here when it approved the modified label. According to MSN, Novartis’s reading, which would allow only for changes relating to different manufacturers, product names, or company addresses, is so narrow that it would “render meaningless the section viii statutory provisions because it would not allow generic companies to carve any language from the brand label in order to avoid brand company patents.” The only way that MSN could have omitted the protected language was to make “minor attendant changes to the label,” and to preclude such modifications “would create a precedent where any brand manufacturer could circumvent section viii statements entirely by carefully wording its indications.”
Both FDA and MSN additionally question whether the carved-out data is actually “critical safety information” as concluded by Novartis. FDA claims that “Novartis overstates [the data’s] significance” while MSN accuses Novartis of asking the Court to “second-guess FDA’s scientific judgment regarding drug safety and efficacy.” As a determination of fact, MSN argues, FDA’s assessment here should be treated with respect even under Loper, which, MSN highlights, clearly states that “Section 706 [of the Administrative Procedure Act] does mandate that judicial review of agency policymaking and factfinding be deferential.” FDA, in responding to the Citizen Petition, laid out a highly reasoned and technical analysis that should be afforded deference, concludes MSN.
At its heart, the question at issue here implicates the survival of the carve-out. While not quite as “on the nose” as the inducement of infringement cases, Novartis is asking the Court to limit FDA’s authority with respect to the carve-out such that the RLD sponsor’s crafting of the indication would dictate whether a carve-out can even be used. This case further raises questions about whether deference will continue to be afforded to FDA in the scientific and factual context notwithstanding Loper. This interesting case is certainly one to watch!